Joe Greener
Improving force fields for biomolecular simulation

Molecular dynamics has shown success in obtaining biological insights by providing mechanistic interpretations of experimental data. However, the force fields used to describe how atoms interact lack accuracy and can fail, for example when applied to disordered proteins or protein aggregation.
We aim to improve force fields for biomolecular simulation in two ways. The first is to use graph neural networks and data from quantum mechanics and experiments to train fast molecular mechanics force fields. The aim is to replace the existing manual approach focussed on specific types of molecules, with a reproducible, easy-to-train model that applies equally to all molecules found in biology.
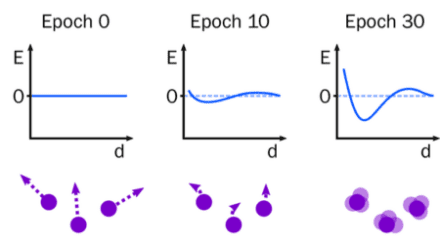
The second approach is to develop machine learning interatomic potentials (MLIPs) that are fast enough to simulate biomolecules. Recently, MLIPs have been developed that rival the accuracy of quantum mechanics, but scaling them to the number of atoms and simulation times relevant to biology has been challenging. We will use simpler models and graphic processing unit (GPU) programming to address this problem, with the eventual aim of simulating enzyme reactions.
The next few decades will see computing resources increase to the point where simulations at biologically relevant length and time scales become routine. We must have accurate physical models available to take advantage of this and to probe the molecular basis of life.