
Pathological filamentous inclusions characterise most human neurodegenerative diseases, including Alzheimer’s and Parkinson’s. Their formation is thought to drive the propagation of inclusions and neurodegeneration. Between 1988 and 1997, we established that the tau protein and alpha-synuclein are the central components of the intracellular amyloid filaments found in most neurodegenerative diseases.
We and others also identified multiplications and mutations in MAPT, the tau gene, that cause inherited forms of frontotemporal dementias with abundant tau inclusions, establishing a central role for tau assembly. SNCA, the alpha-synuclein gene, is multiplied or mutated in Parkinson’s disease, dementia with Lewy bodies and juvenile-onset synucleinopathy.
In collaboration with Sjors Scheres, we are using cryo-EM to determine the structures of pathological amyloid filaments made of assembled tau, alpha-synuclein, amyloid-beta and TMEM106B. This has led to a structure-based classification of human neurodegenerative diseases.
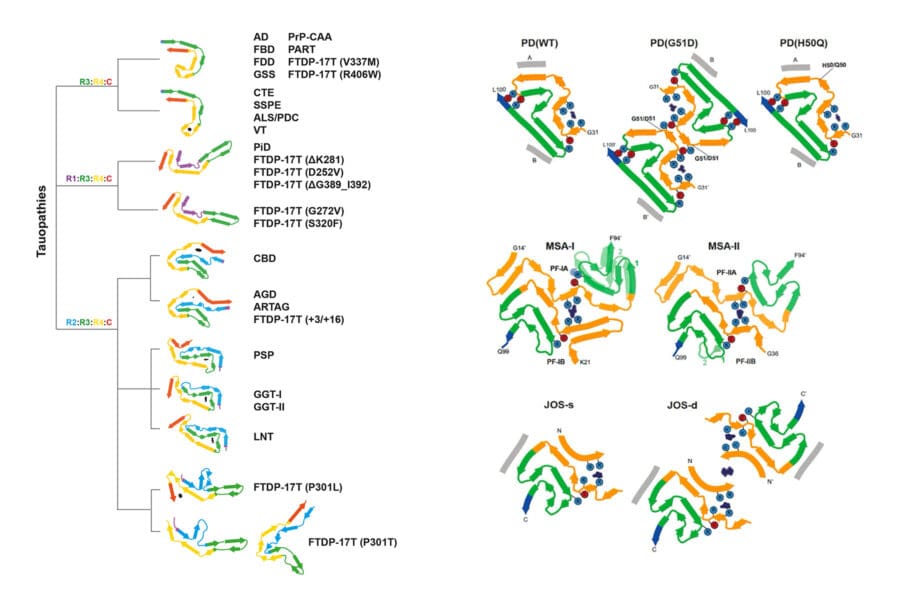
To understand mechanisms of disease, we must develop methods for forming amyloid filaments like those from human brains. We anticipate that this will result in improved diagnostics and the development of generally effective mechanism-based therapies. Until recently, none of the known structures of tau and alpha-synuclein filaments assembled from recombinant proteins were the same as those from the human brain. However, we have now been able to show that recombinant tau (297-391) assembles into filaments that are identical to the paired helical filaments of Alzheimer’s disease and other conditions. The same is true of full-length human tau, in which 12 serine/threonine residues were mutated to aspartate.